The Motherhood Space
This space is designed to help new and expectant moms who are experiencing maternal mental health challenges by improving their ability to cope and function in daily life.

Jacksonville’s Most Preferred Health Care
This space is designed to help new and expectant moms who are experiencing maternal mental health challenges by improving their ability to cope and function in daily life.

Safely providing you and your family with high-quality care is what we do every day.

Our new portal provides secure access to your medical information anytime, anywhere. View test results, make appointments and much more for you and the people you care for.

Seniors in our community are struggling with hunger at twice the national rate. We're helping address this challenge by donating food from our cafeterias.

Let us help you decide. Get a virtual evaluation from the comfort of home, or wherever you are, with one of our emergency-trained physicians. To expedite your visit, create an account now.

If you want to see a doctor but aren’t sure who to call, let our helpful care coordinators get you an appointment with the right doctor for you. Simply call 904.202.4YOU (4968) or fill out our short appointment request form.

Get free, easy-to-use tools and resources to help you start conversations with kids and teens, boost their mental well-being and break stigmas associated with children’s mental health.
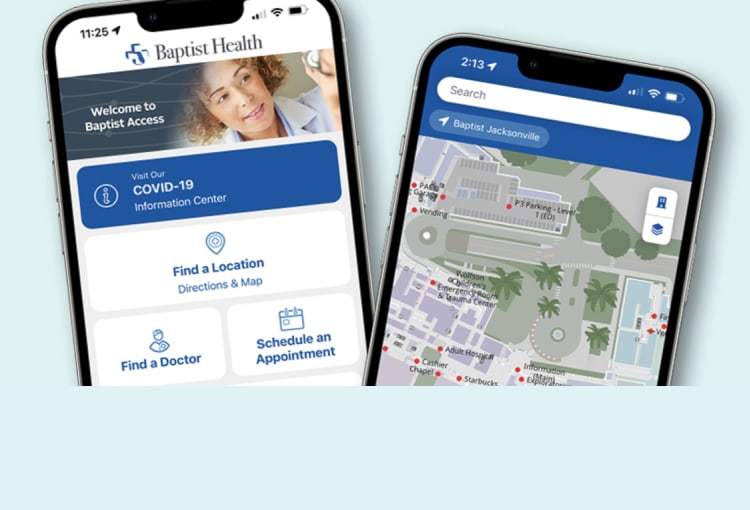
Whether you need to find a location in the hospital or find a doctor near you, you can do it all from your phone with Baptist Access.
News, information, tips and inspiring stories written by Baptist Health experts to improve your health and wellness.